

AML remains a therapeutic challenge with a high mortality rate, underscoring the need for new biologically based therapies. Somatic alterations that deregulate epigenetic programs and signal transduction pathways frequently coexist in AML. While the former class of alterations is hypothesized to promote a chromatin state that is permissive for AML development and essential for leukemia maintenance, experimental data also suggest that signaling mutations play a central role in driving leukemic growth. Thus, simultaneously targeting the abnormal epigenetic landscape and aberrant signaling is a rational therapeutic approach. We previously used the MEK inhibitor PD0325901 (PD901) to target the Raf/MEK/ERK (MAPK) effector pathway in primary murine AMLs in vivo. Here we report substantial preclinical efficacy of the BET inhibitor PLX51107 in several independent primary AMLs that was further enhanced by PD901.
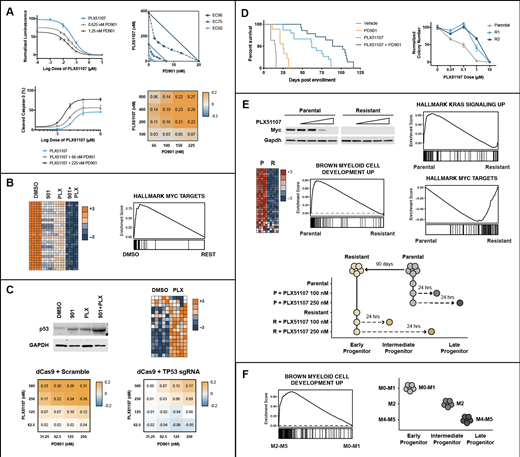
We first exposed five NRAS or KRAS mutant human AML cell lines to varying concentrations of PLX51107 and PD901. These studies revealed potent synergy based on cell proliferation and apoptosis readouts (Figure 1A). We next performed RNA-seq on the NRAS-mutant OCI-AML3 cell line treated with PD901, PLX51107, or the combination. Remarkably, PLX51107 + PD901 cooperated to potently downregulate MYC and its transcriptional targets (Figure 1B). Additionally, AML cells increased the expression of p53 transcriptional targets in response to PLX51107 and CRISPRi-mediated knock-down of TP53 expression abrogated the synergistic effects of PLX51107 + PD901 (Figure 1C).
We previously performed retroviral insertional mutagenesis in NrasG12D mice to generate genetically diverse panels of transplantable, primary AMLs. To follow-up on our promising in vitro data, we transplanted 5 of these leukemias into cohorts of recipient mice and treated them with PLX51107 ± PD901. PLX51107 (10 mg/kg/day) showed impressive efficacy that was further enhanced by a modest dose of PD901 (1.5 mg/kg given 4 days per week; Figure 1D).
To begin characterizing mechanisms of resistance, we evaluated matched pairs of drug-sensitive ("parental") and relapsed leukemias that emerged during drug treatment. We selected relapsed leukemias that exhibited clonal evolution based on distinct novel retroviral integrations for in depth evaluation. Re-transplanting these AMLs into secondary recipients and re-treating them with PLX51107 ± PD901 confirmed intrinsic drug resistance, which was further validated ex vivo in dose response methylcellulose assays (Figure 1D). Remarkably, several of these resistant leukemias down-regulated basal Myc protein expression, which supports the idea that their transcriptional networks are "rewired" (Figure 1E).
We next compared basal and dynamic transcriptional profiles in parental and resistant leukemias by exposing them to PLX51107 ex vivo and then performing RNA-seq analysis. Intriguingly, PLX51107 treatment of parental AMLs resulted in transcriptional changes consistent with myeloid maturation in a dose dependent manner (Figure 1E), suggesting that BET inhibition induces differentiation in these leukemias. By contrast, paired resistant AMLs are relatively "immature" compared to their parental counterparts (Figure 1E), which supports this idea that in vivo treatment with PLX51107 selects for the outgrowth of clones at an earlier progenitor state. These resistant AMLs also upregulate Myc transcriptional targets and unexpectedly downregulate Ras transcriptional targets (Figure 1E). We corroborated these findings by analyzing large pediatric AML transcriptome datasets generated as part of the NCI TARGET initiative (Figure 1F). Taken together, our data support the hypothesis that BET inhibitor and MEK inhibitor combination may be a particularly effective therapeutic approach in AML. Additional preclinical testing in patient derived xenograft models is underway.
Burgess:Bristol Myers Squibb: Current Employment. Severson:Plexxikon Inc.: Current Employment. Powell:Plexxikon Inc.: Current Employment. Bollag:Plexxikon Inc.: Current Employment.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal